Medical Device Software Development: Key Trends, Compliance, and Best Practices

Vishal Panchal
The global medical device market is bound to reach $571.72 billion in 2024. It is forecast to exceed $717.38 billion by 2030. That growth reflects something deeper. It shows an increasing demand for medical devices and software that enable deeper insights for healthcare service providers.
Hospitals, diagnostics platforms, surgical robotics systems, and wearable manufacturers are all demanding connected, intelligent, regulation-ready software at a pace that shows no sign of slowing. The challenge is that medical device software development is not a standard engineering problem.
Patient safety, regulatory scrutiny, and clinical liability intersect at every architectural decision. A bug in a consumer app can cost a healthcare service provider or hospital millions. And on top of it all, a simple failure in a connected insulin pump or radiation therapy control system costs a life.
So, there is no denying that, as a healthcare service provider, hospital, or medical device manufacturer, a software component is key. But how do you ensure you develop the right software? This guide will help you determine why you need medical device software, its compliance requirements, trends, and best practices.
Why Medical Device Software Development Demands a Higher Engineering Standard?
Standard software engineering tolerates iteration, retrospective documentation, and informal acceptance criteria. Medical device software development operates under a fundamentally different contract with failure. Every unverified edge case is a regulatory exposure. Every undocumented design decision is an audit finding waiting to surface.
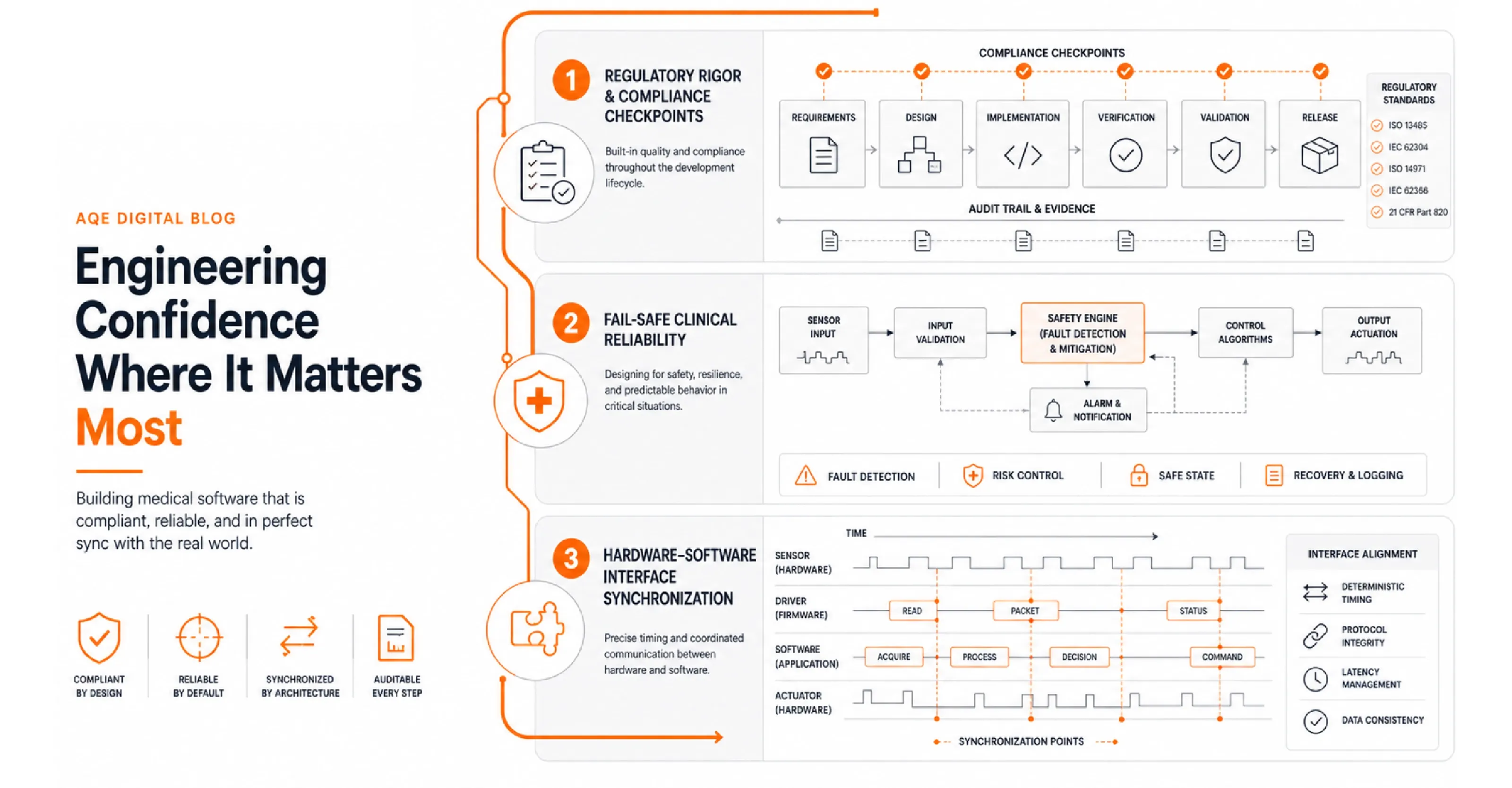
Teams that underestimate the rigor required for developing medical software pay for it downstream: delayed submissions, complete redesigns, or post-market field corrections that cost far more than the original development budget.
Medical device software development programs that fail at submission almost always share the same root cause: compliance was treated as a documentation layer bolted onto the end of engineering, rather than a process discipline woven into every sprint from day one.
Clinical Software Development: Where Fail-Safe Is a Safety Constraint
Clinical software development does not treat reliability as a quality attribute. It treats it as a hard safety constraint.
IEC 62304 classifies medical device software into three safety classes: Class A (no injury possible), Class B (non-serious injury), and Class C (serious injury or death). Class C requires full lifecycle documentation, formal change control, and end-to-end traceability from design inputs through to verification evidence.
Teams that classify at Class B and later identify a Class C failure mode do not patch a module. They rebuild their entire quality management infrastructure. The cost of misclassification is not a documentation gap. It is a project restart.

Medical Software Design and the Hardware-Software Interface
Medical software design does not operate in isolation from the physical device it controls. Software can iterate in two-week agile sprints. Hardware fabrication and tooling cycles run four to six weeks. Teams that treat firmware as just another software layer collide with this mismatch on every release.
Software development for medical devices that ignores this dynamic produces integration failures that are expensive to diagnose and even more expensive to resolve under regulatory scrutiny. Define hardware-software synchronization points explicitly in the SDLC, align sprint cadences with hardware lead times, and lock interface specifications before either side enters deep implementation.
What Are the Core Compliance Frameworks for Medical Software?
Healthcare software compliance is not a final documentation exercise. Both the FDA and the EU MDR expect compliance posture to be embedded in the development process from day one.

- IEC 62304 defines the lifecycle requirements for medical device software, covering architecture, coding, verification, maintenance, and problem resolution.
IEC 62304 Edition 2, expected in 2026, introduces explicit requirements for AI and machine learning development. It also simplifies the safety classification framework, moving toward a two-class system.
- ISO 13485 governs the quality management system surrounding the development process, including document control, supplier qualification, and post-market surveillance infrastructure.
As of February 2026, the FDA's new Quality Management System Regulation formally incorporates ISO 13485:2016 by reference, replacing the legacy Quality System Regulation. Programs still running on the old QSR now carry an active compliance gap.
- The FDA's SaMD Framework applies to Software as a Medical Device, where software performs a clinical function independent of physical hardware.
- SaMD programs must comply with the FDA's premarket cybersecurity guidance, which requires threat modeling, a Software Bill of Materials, and a documented vulnerability management plan to be submitted with every 510(k) or De Novo application.
Healthcare software compliance failures carry consequences that extend far beyond regulatory delay. The average U.S. medical device recall costs more than $600 million in corrective actions, field notifications, and reputational damage.
Getting compliance architecture right at the start of medical device software development is not overhead. It is risk elimination.
What Trends Are Shaping Medical Device Software in 2026?
Software development for medical devices is accelerating across four dimensions. Each one adds engineering complexity and regulatory obligation:
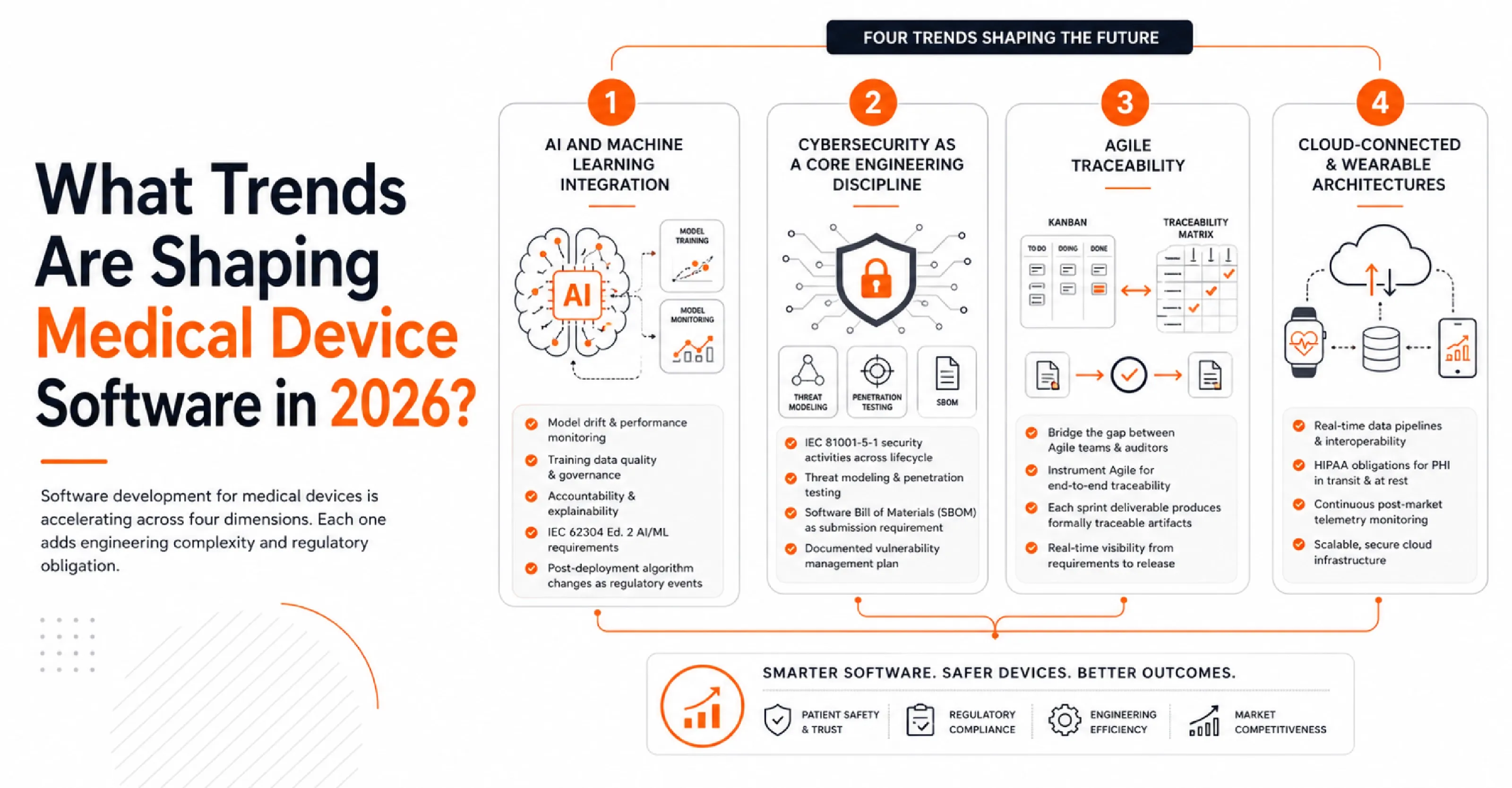
- AI and Machine Learning Integration: According to FDA tracking data, the agency authorized more than 1,400 AI and ML-enabled medical devices by the end of 2025. AI introduces model drift, training data dependency, and accountability gaps that deterministic software does not create.
Teams must satisfy IEC 62304 Edition 2's incoming AI/ML requirements, and the FDA's SaMD action plan, which treats post-deployment algorithm changes as distinct regulatory events.
- Cybersecurity as a Core Engineering Discipline: Connected medical devices are high-value attack targets. The FDA now recognizes IEC 81001-5-1 as the standard for security activities across the healthcare product lifecycle.
Threat modeling, penetration testing, and a documented Software Bill of Materials (SBOM) are submission requirements, not optional safeguards.
- Agile Traceability (The Documentation Paradox): Regulated software engineering teams face a tension where developers want Kanban boards, while auditors want linear traceability matrices.
The answer is not to abandon Agile, but to instrument it so that every sprint deliverable produces formally traceable artifacts.
- Cloud-Connected and Wearable Architectures: According to Grand View Research, the global smart medical devices market was valued at $90.5 billion in 2024 and is projected to reach $185.6 billion by 2030.
Developing clinical software for these platforms adds real-time data pipeline requirements, HIPAA obligations for protected health information in transit, and continuous post-market telemetry monitoring. Strong Medical Device Engineering services and practices are essential here, ensuring security, interoperability, regulatory compliance, and system reliability are built into the product lifecycle from design through deployment.
What are the key Medical Device Software Engineering Best Practices?
Medical device software engineering demands a structured process discipline that would be overkill in consumer software and non-negotiable in regulated environments. These practices consistently separate compliant programs from those that stall.
1. Classify Before You Architect.
IEC 62304 classification determines your process depth. Running medical device software development at Class B discipline when your actual failure modes require Class C means rebuilding your entire documentation infrastructure mid-project. Classification is the first engineering decision, not the last.
2. Build a Requirements Traceability Matrix from Day One.
Every requirement must link to a design specification, a test case, and a verification result. Medical device software development teams that build traceability retroactively consistently hit the same wall: disconnects between what was designed, built, and tested that cannot be reconciled without returning to requirements. A gap in the RTM is grounds for rejection of an FDA submission.
3. Apply ISO 14971 Risk Management in Parallel.
Medical device software development programs that defer risk management until the verification phase are solving the wrong problem at the wrong time. Every hazardous situation in the risk file must trace to a specific software mitigation in the design controls before architecture is locked.
For clinical software development, each sprint does not close until verification evidence exists for the features delivered and not deployed, and they are verified, documented, and traceable.
Regulatory clearance is not the endpoint. Software development for medical devices under the EU MDR and FDA frameworks requires a post-market surveillance plan that covers anomaly reporting timelines, corrective action procedures, and version control discipline to support audit readiness throughout the product's commercial life.

Conclusion
Medical device software development is where engineering rigor and patient safety converge under regulatory authority. The compliance bar is not staying flat. IEC 62304 Edition 2, the FDA QMSR, and SaMD cybersecurity requirements are active constraints on programs running today, not future considerations.
Medical device software development done right means classification before architecture, compliance embedded in every sprint, and post-market surveillance designed before the first line of code ships. The same discipline becomes even more critical when building Clinical AI Solutions, where patient safety, traceability, validation, and regulatory readiness must align from day one. Done wrong, it means recalls, submission failures, and corrective action cycles that cost multiples of the original program budget.
The path to clearance runs through a disciplined engineering process, not heroic late-stage documentation.